Pooled CRISPR Technologies
We help collaborators in experimental design and execution, choosing the best model system, perturbation type, challenge, and readout. Our tools include CRISPR knockout, interference, base editing, CRISPRoff, and ORF overexpression.
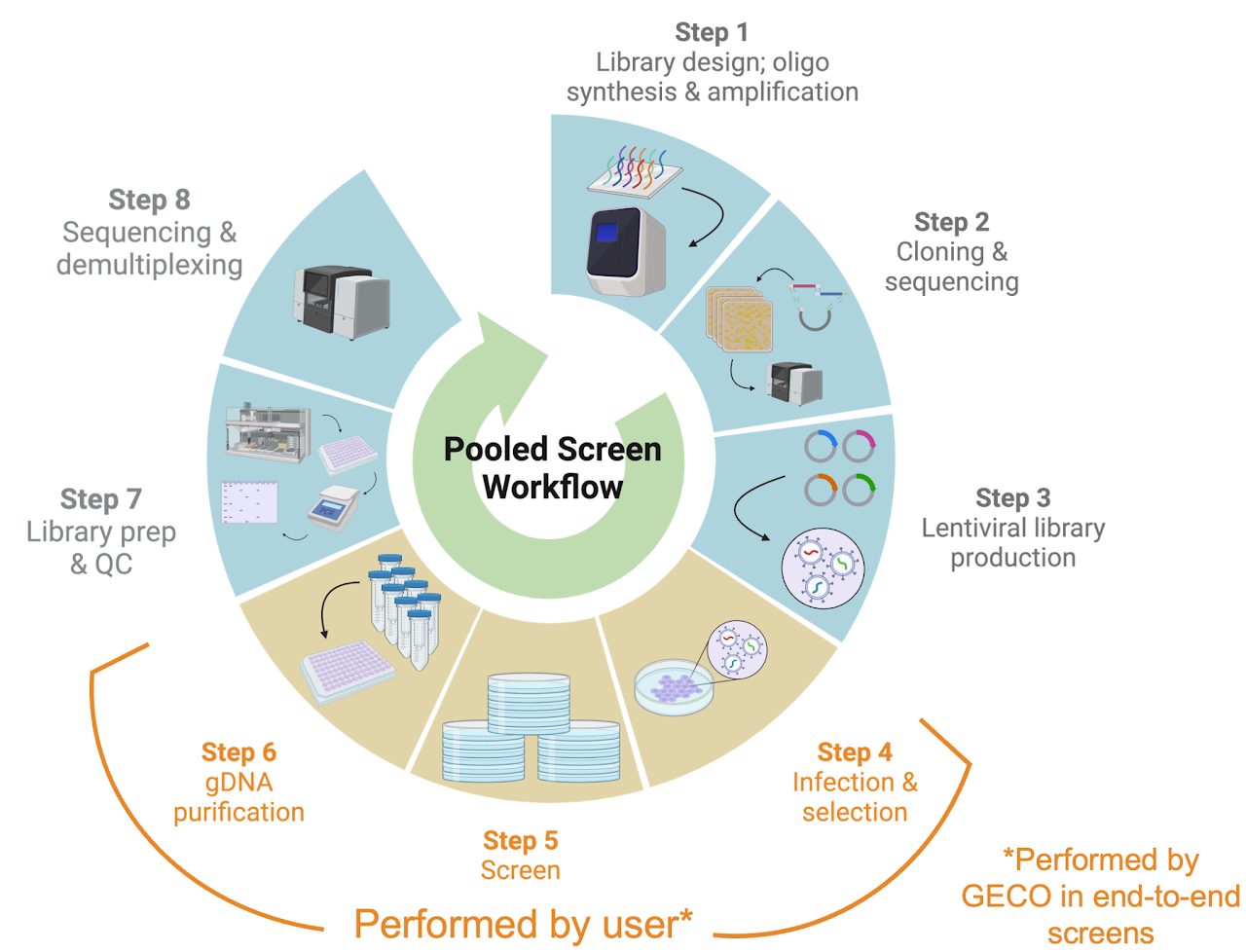
Experimental Design
Every pooled screen is defined by four dimensions. We work with you to choose the right combination for your biological question.
The Model
Cell lines, primary cells, organoids, or in vivo models.
The Perturbation
CRISPR KO, CRISPRi, CRISPRa, CRISPRoff, base editing, or ORF overexpression.
The Challenge
Proliferation, drug resistance, pathogen exposure, differentiation, or other selective pressures.
The Readout
sgRNA or amplicon sequencing, scRNA-seq (Perturb-seq), multi-omics, FACS, or imaging.
CRISPR Knockout Screens
The in4mer Platform
The in4mer CRISPR/enAsCas12a multiplex knockout platform uses arrays of four independent guide RNAs targeting the same or different genes, delivered via an all-in-one lentiviral vector. The enAsCas12a nuclease (Kleinstiver et al., 2019) offers several advantages over Cas9: it uses shorter guide RNAs (~40 nt vs ~100 nt), has a different PAM requirement (TTTV) that expands targeting space, and enables efficient multiplex knockout from a single transcript. The in4mer system was developed by the Hart and Doench labs and published in Anvar et al. (2024).
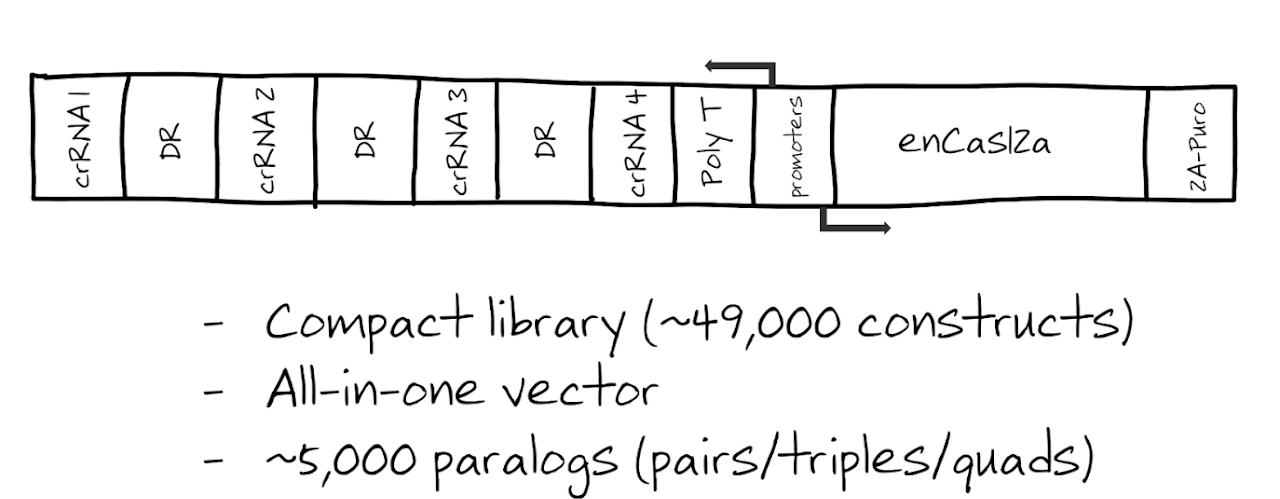
The Inzolia Library
The genome-scale Inzolia library targets ~19,700 single genes and covers >4,900 paralog combinations (pairs, triples, and quads). It is 30% smaller than typical CRISPR/Cas9 libraries while maintaining high sensitivity in identifying core and context-dependent essential genes, effectively detecting synthetic lethal and buffering genetic interactions between paralogs (Anvar et al., 2024).

What ATG-C Provides
- Build or purchase pDNA libraries
- Amplify and validate libraries (Sanger, long-read and short-read NGS)
- Generate lentiviral libraries, including concentrating liter-size lentiviral preps for all-in-one vectors
- Conduct functional batch testing in cancer cell lines
- Perform library prep, QC, and sequencing of submitted samples
Your Responsibilities
- Cell line optimization (lentiviral titering, puromycin kill curves, polybrene testing)
- Transduction of viral library
- Maintain ≥10 cell divisions post-transduction
- Collect dry cell pellets and store at −80°C
- Perform lysis and gDNA isolation
- Submit gDNA and plate map to ATG-C
Proliferation Screen Example
For a genome-wide proliferation screen using the Inzolia library (~19,700 genes + 4,900 paralog combos), typical parameters include:
- Library size: ~25,000 constructs
- Coverage: 500–1,000× per construct
- Cells needed: 12.5–25 million transduced cells per replicate
- gDNA requirement: ~200 μg per sample (at 6.6 pg/cell)
Use our in4mer screen size calculator to estimate cell numbers, gDNA requirements, and sequencing depth for your experiment.
CRISPRi / CRISPRoff / CRISPRa
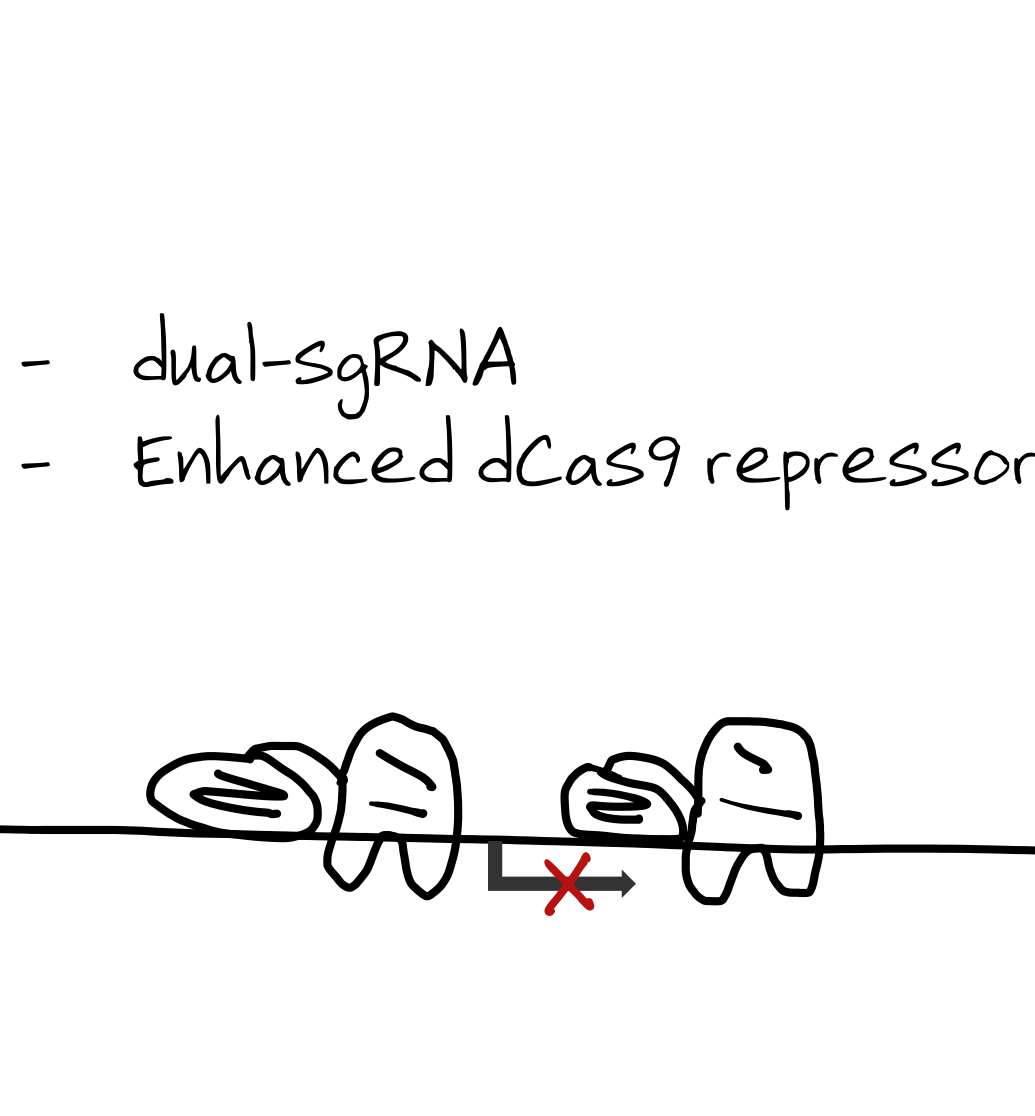
CRISPRi (CRISPR Interference)
A catalytically dead Cas protein (dCas9) fused to repressor domains is targeted to a promoter or enhancer, creating a steric block that transiently inhibits transcription. We use functionally validated genome-wide CRISPRi libraries with dual-guide constructs and enhanced CRISPR repressors to minimize off-target silencing and toxicity (DeWeirdt et al., 2021).
CRISPRoff
dCas9 fused to both repressor and methylase domains targets gene promoters. Genes with CpG islands can be durably repressed — epigenetic repression that persists in daughter cells. This enables heritable gene silencing without DNA cutting.
CRISPRa (CRISPR Activation)
dCas9 fused to activator domains recruits transcriptional machinery to a promoter, enabling gain-of-function screens at genome scale.
Base Editing
Nickase Cas (nCas9 or nCas12a) fused to a deaminase causes C→T, A→G, or C→G conversions without double-strand breaks. Used for targeted saturation mutagenesis, allele correction, and functional variant screens.
Perturb-seq
Perturb-seq combines CRISPR-mediated perturbations with single-cell RNA sequencing. Direct-capture technology detects which perturbation(s) occurred in each cell by capturing the sgRNA sequence. These screens are typically short (1–8 days post-transduction). The approach was pioneered by multiple groups simultaneously (Dixit et al., 2016; Adamson et al., 2016; Jaitin et al., 2016; Datlinger et al., 2017).
We have had excellent results using enhanced dCas9 repressors and dual-sgRNA vectors. The technology allows for up to genome-wide RNA-seq profiling in a single experiment.
Additional readout modalities include FACS-based screens for surface marker or reporter expression, and ORF overexpression screens for gain-of-function phenotyping. For a comprehensive review of pooled CRISPR screen design, see Bock et al. (2022).
How to Start a Project
-
Schedule a planning meeting
Optional for collaborators with a ready-to-screen library. Recommended for new projects to discuss model system, perturbation type, and experimental design.
-
Order the experiment on iLab
Submit your request through iLab.
-
Fill out the project description form
Provide details on cell line, perturbation type, challenge conditions, and readout.
-
Schedule a pickup
Bring a dry ice container for virus transport.
-
Return gDNA
After completing the screen, submit your gDNA samples and plate map for library prep and sequencing.
References
- Anvar et al. (2024) Nature Communications. DOI: 10.1038/s41467-024-47795-3
- Bock et al. (2022) Nat Rev Methods Primers. DOI: 10.1038/s43586-022-00098-7
- Kleinstiver et al. (2019) Nature Biotechnology. DOI: 10.1038/s41587-018-0011-0
- DeWeirdt et al. (2021) Nat Biotechnol. DOI: 10.1038/s41587-020-00745-6
- Datlinger et al. (2017) Nat Methods. DOI: 10.1038/nmeth.4177
- Dixit et al. (2016) Cell. DOI: 10.1016/j.cell.2016.11.038
- Adamson et al. (2016) Cell. DOI: 10.1016/j.cell.2016.11.048
- Jaitin et al. (2016) Cell. DOI: 10.1016/j.cell.2016.11.039